Introduction¶
MolHash is a command-line application and programming library for generating hashes from molecular structures. This section gives an overview of each of the most useful hash functions in turn. The user should find it straightforward to add additional hash functions, or tweak the existing ones.
To begin with, the following table summarises the available molecular hashes, whether their calculation alters the structure (this is only relevant to API use, not via the command-line), and whether they are sensitive (or invariant) to the presence of particular molecular features. If a particular hash function is sensitive to a molecular feature but this is not desired, then the molecule should be normalized accordingly with the provided normalization methods/options.
| Hash functions | May alter structure | Sensitive to | ||||
|---|---|---|---|---|---|---|
| Explicit H | Isotope | Atom class | Atom stereo | Bond Stereo | ||
| Anonymous Graph | Y | Y | Y | Y | Y | N |
| Arthor Substructure Order | N | Y | Y | N | N | N |
| Atom & Bond Counts | N | Y | N | N | N | N |
| Canonical Smiles | N | Y | Y | Y | Y | Y |
| Degree Vector | N | Y | N | N | N | N |
| Element Graph | Y | Y | Y | Y | Y | N |
| Heteroatom Protomer | Y | Y | Y | Y | Y | N |
| Heteroatom Tautomer | Y | Y | Y | Y | Y | N |
| Mesomer | Y | Y | Y | Y | Y | N |
| Molecular Formula | N | N | N | N | N | N |
| Murcko Scaffold | Y | Y | Y | Y | Y | Y |
| Extended Murcko | Y | Y | Y | Y | Y | Y |
| Net Charge | N | N | N | N | N | N |
| Redox Pair | Y | Y | Y | Y | Y | N |
| Regioisomer | Y | Y | Y | Y | Y | Y |
| SmallWorld Index BR | N | Y | N | N | N | N |
| SmallWorld Index BRL | N | Y | N | N | N | N |
Anonymous Graph¶

This is the canonical SMILES string for a molecule after setting all of its atoms to asterisks, and its bonds to single bonds. It can be used to identify molecules that have the same graph structure independent of atom identity, bond order or hydrogen count.
Arthor Substructure Order¶
Arthor is a substructure/similarity search engine developed by NextMove Software. When performing a substructure search, results are returned in the order in which they are present in the database. If users sort their database entries by this hash, results will be returned based in an order that approximates similarity to the query but favoring ‘plain’ molecules.
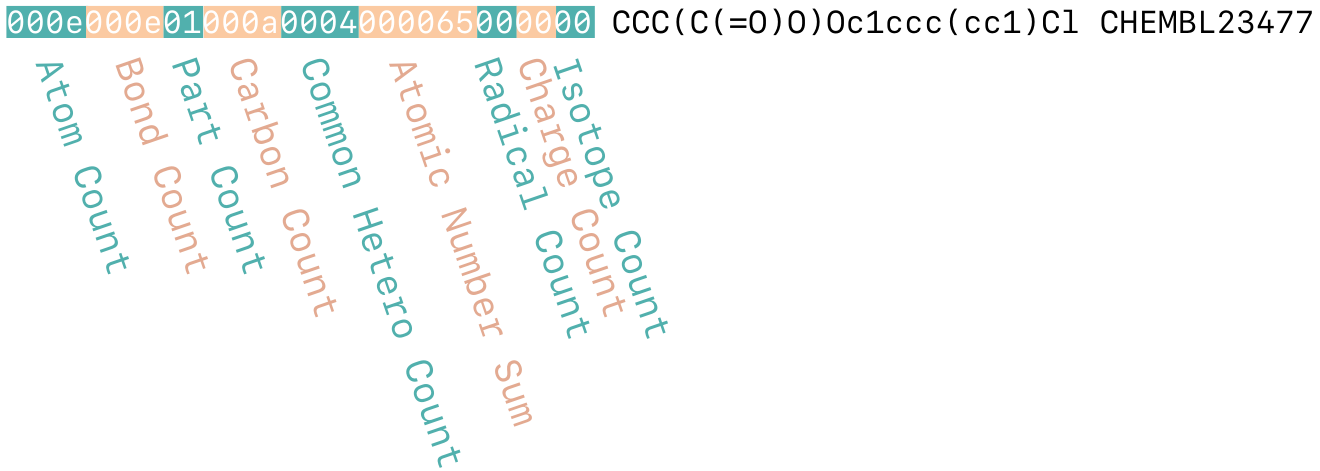
Canonical SMILES¶
Generate a canonical SMILES string that includes each of the following (if present, and if the toolkit supports its inclusion): atom and bond stereo, atom maps, explicit hydrogens and isotopes.
Element Graph¶

This is the canonical SMILES string for a molecule after settings all its bonds to single bonds and normalizing hydrogen counts. It can be used to identify molecules that have the same bonding arrangement but different bond order.
Heteroatom Protomer/Tautomer¶
These molhashes are minor variations of each other, and consist of a SMILES component followed by one or two integers. The SMILES is a canonical SMILES generated after stripping all hydrogens and charges, and setting all bond orders to 1. The tautomer variant appends two integers, the total number of hydrogens on non-carbon atoms, and the total charge; the protomer variant subtracts the charge from hydrogen count to yield a single value.



To illustrate their use and their difference, consider the structures above on the left-hand side, which all hash to the same SMILES component (depicted on the right). These three all have the same protomer hash (C[C]1[CH][N][CH][N]1_1) but because they have different total charges, the tautomer hash shared by the first two (C[C]1[CH][N][CH][N]1_1_0) is different for the third (C[C]1[CH][N][CH][N]1_2_1). In other words, the protomer variant may be used to find tautomers independent of the charge state.
Mesomer/Redox Pair¶
These two molhashes are minor variations of each other, and consist of a SMILES component which is followed by a single integer in the case of the mesomer hash. The SMILES is a canonical SMILES generated after setting all charges to zero and bond orders to 1. The mesomer variants appends the total charge to the hash.


The example above shows two redox pairs. These have the same redox pair hash ([CH]1[CH][CH][C]2[C]([CH]1)[N][C]([C]([N]2)[O])[O]) but different overall charge and hence have different mesomer hashes (the first has _-2 appended, while the second has _0 appended).


As an example of a mesomeric pair, consider the charge-separated and hypervalent forms of nitros above. These have the same mesomer hash (CCN([O])[O]_0).
Molecular Formula¶
The molecular formula is possibly the most widely used molecular hash, a concise description of the atomic composition and formal charge, that excludes information on bonding. The molecular formula can be used to identify isomers, constitutional or otherwise.
Murcko Scaffold and Extended Murcko¶
The Murcko scaffold hash is the canonical SMILES of a molecule after removing all substituents.

The extended Murcko hash is the canonical SMILES of a molecule after replacing all substituents with attachment points (an asterisk in SMILES).

Regioisomer¶
The regioisomer hash is the canonical SMILES for a molecule after breaking a subset of acyclic single bonds and replacing the connection by an asterisk or hydrogen. Specifically, acyclic single bonds are cut if either end of the bond is involved in a ring or if the bond is between a non sp2-hybridized carbon atom and a non-carbon atom.
